Research
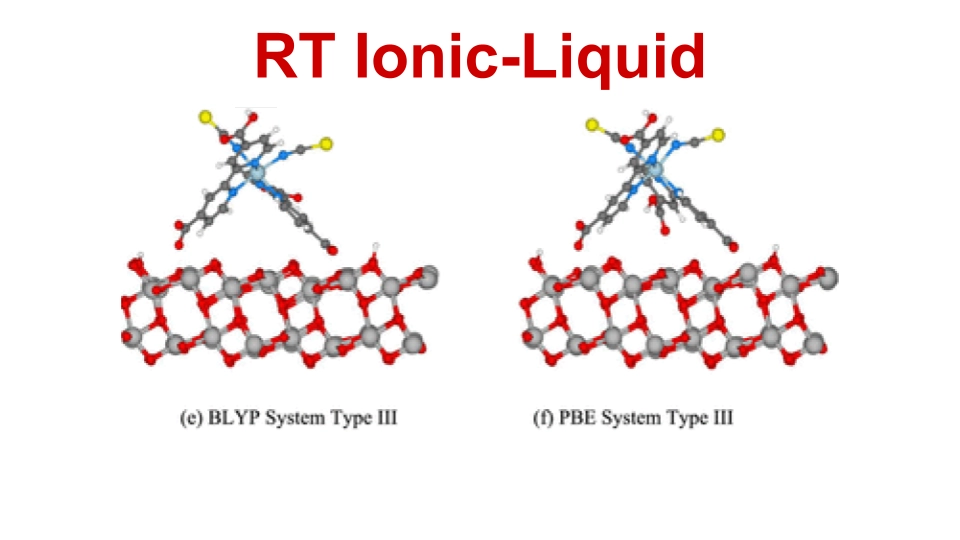
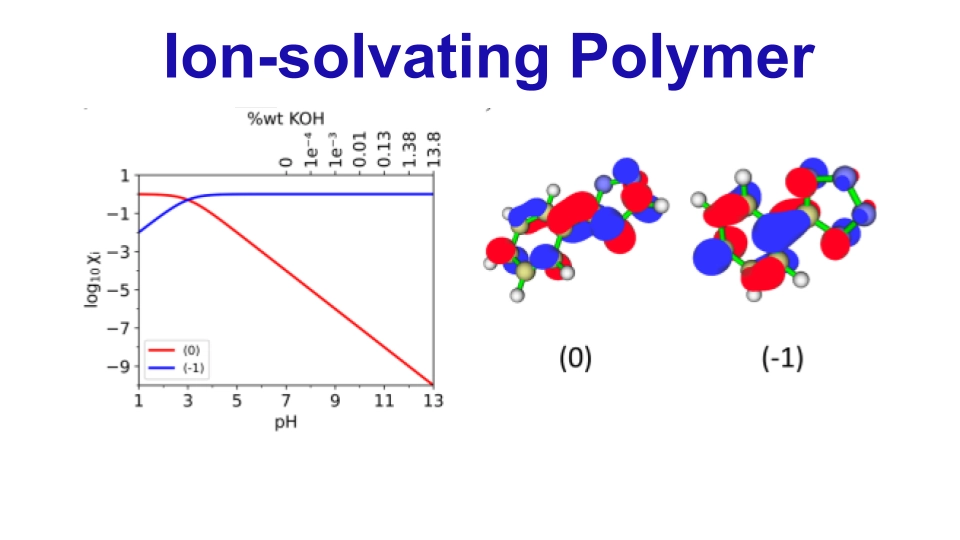
Technical University of Denmark (DTU)
Nonequilibrium Dynamics with AI/ML potentials at the Solid-Liquid Interface
We looked into the O2 reduction by protons and alkali metal ions in a nonaqueous electrolyte, like acetonitrile, to understand the effect of the solvents. The Oxygen Reduction Reaction (ORR) is highly fascinating in relation to possible future lithium- and sodium-air batteries, which have incredibly huge theoretical capacities. The ORR selectivity in alkali metal-air batteries for superoxides, peroxides, or dioxides is influenced by the cathode material, the electrolyte solvent, and the trace water content. The capacity and rechargeability of batteries are subsequently impacted by these discharge products. Specifically, superoxide dissolution is necessary to boost battery capacity, and it has been demonstrated that trace water contaminants aid in this dissolution.
University College Dublin (UCD)
Molecular simulation of energy-transfer processes at interfaces
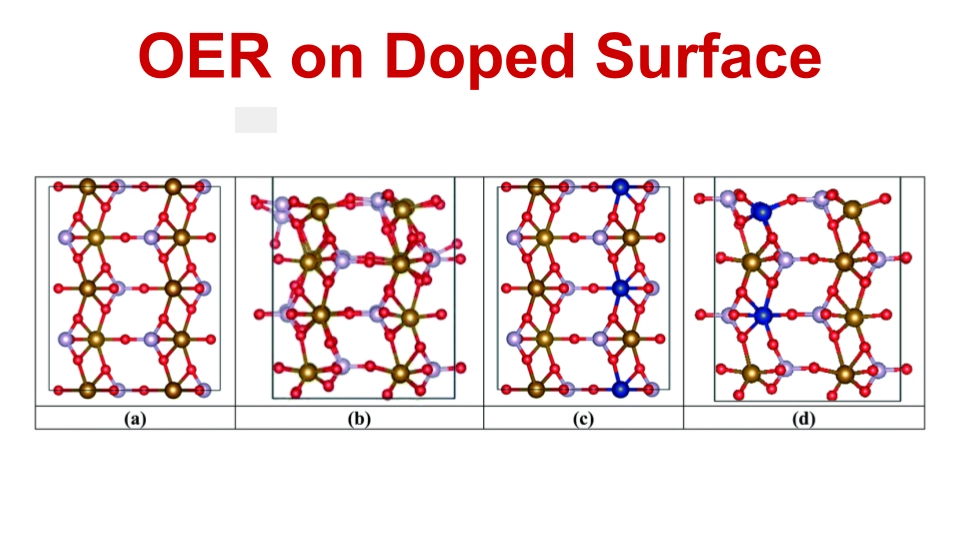
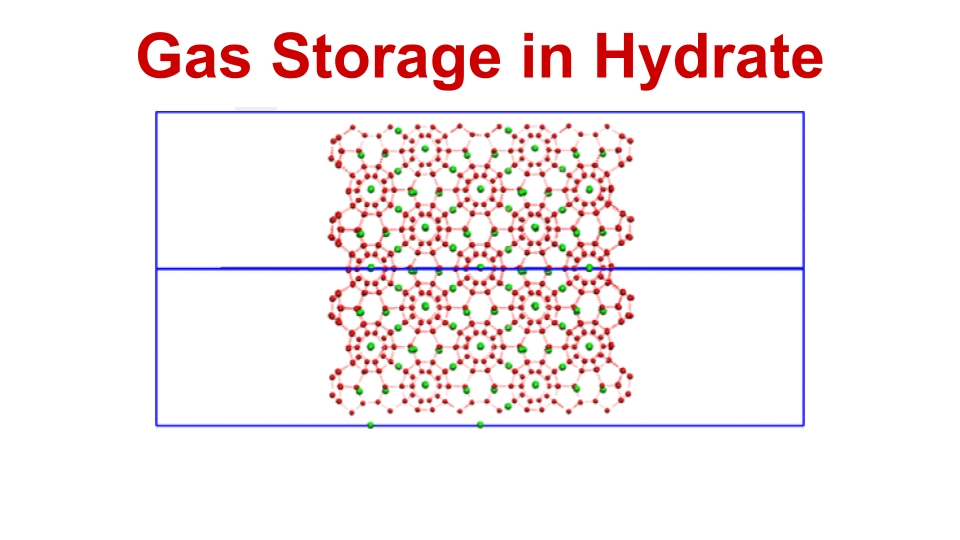
We investigated the complex clathrate hydrate interfaces for gas storage applications and, for my thesis work, we also investigated doped iron phosphate materials for water splitting reactions. Explore the study of sII type hydrate structures in clathrate hydrates containing neon, hydrogen, and deuterium gases. Using first-principles calculations, the oxygen-evolution reaction (OER) pathway of water on a doped Fe3Co(PO4)4 surface was computed.
Indian Institute of Technology Jodhpur (IITJ)
Fabrication and Analysis of Gas-Sensing Devices
We utilized Chemical Vapour Deposition (CVD) and molecular-beam epitaxy (MBE) techniques for the fabrication of gas-sensing devices. We analyzed and tested the thin film gas-sensing unit.
Theoretical reaction dynamics and rate theories, Direct dynamics simulations
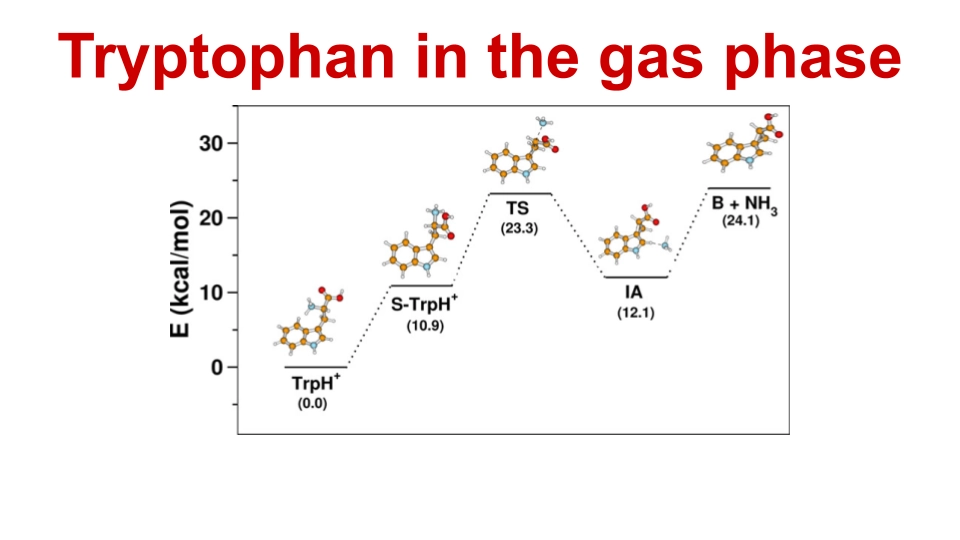
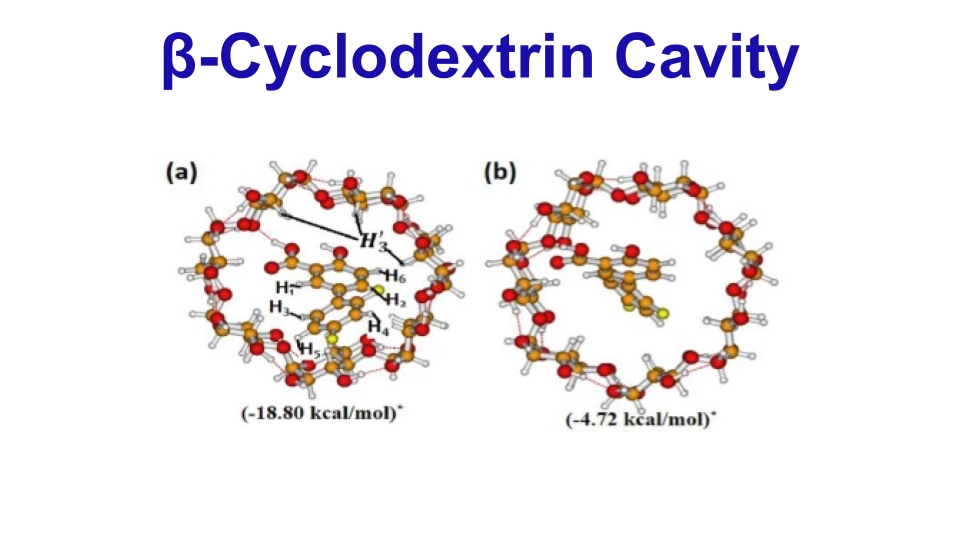
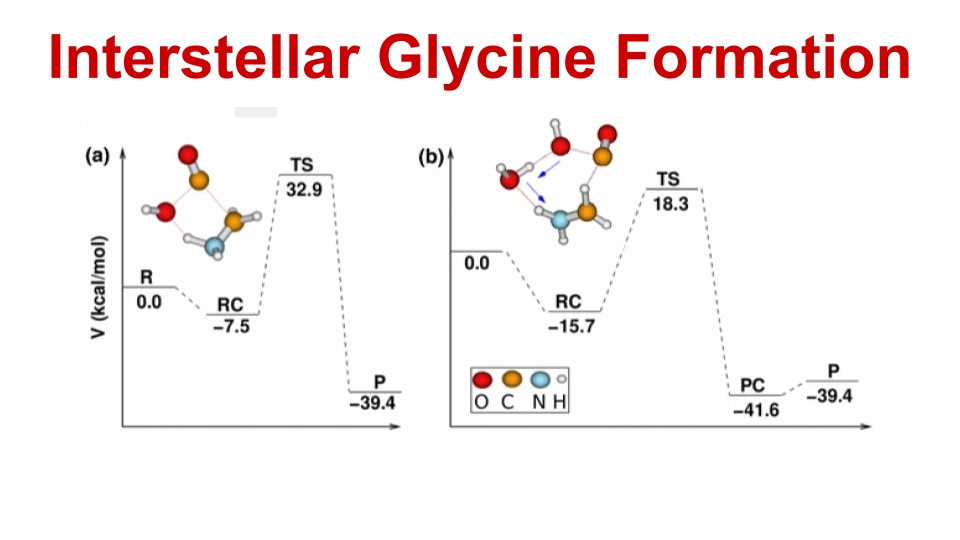
We studied the decomposition pathways of protonated tryptophan using Density Functional Theory (DFT) and Ab initio molecular dynamics (AIMD) simulations. We explored how simple organic molecules may have formed in space and traveled to Earth on meteorites, potentially leading to the origin of life. In addition, we collaborated with NMR experimental researchers to calculate the minimum energy geometry of inclusion complexes.
PSG College of Technology, India (PSG)
Thin film thermoelectric materials
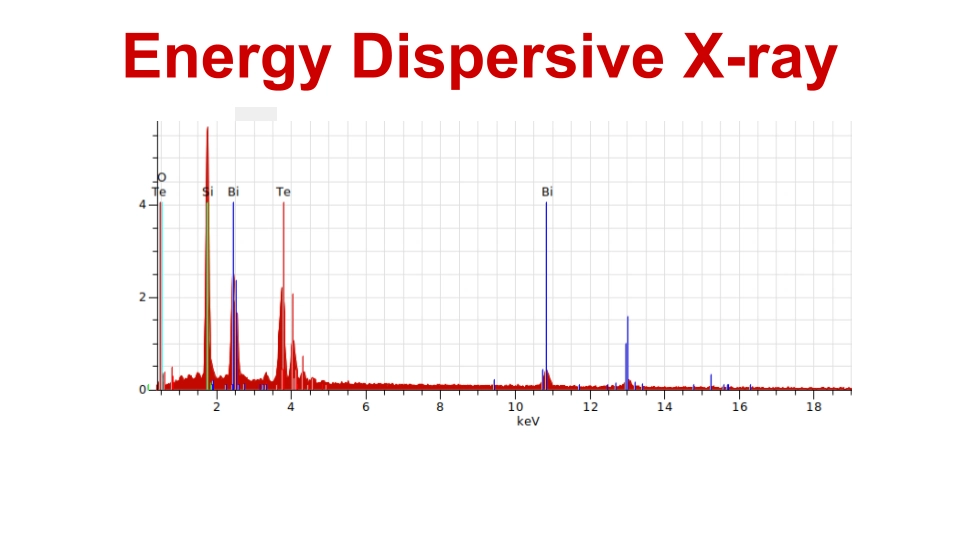
Bismuth telluride thin films were fabricated by electron beam evaporation unit. X-ray diffraction (XRD) patterns and XPS analyses were performed on both as-deposited and post-annealed bismuth telluride films. We tested the Bi2Te3 thin films to determine their suitability for thermoelectric applications.
Selected Publications
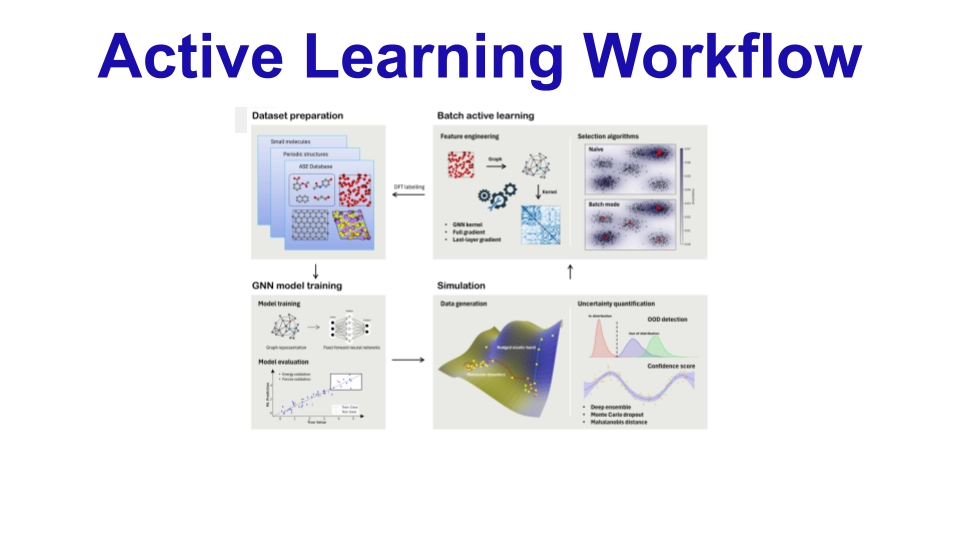
CURATOR represents a significant advancement in the field of computational materials science by enabling the efficient and rapid development of high-accuracy Machine-Learned Interatomic Potentials (MLIPs). Its integration of state-of-the-art models, uncertainty quantification, and novel gradient computation methods significantly reduces the resources and time required for MLIP development. The promising results in complex material applications highlight CURATOR's potential in accelerating materials discovery. More extensive and longer time-scale atomistic simulations are thereby made possible by the flexibility and efficiency of CURATOR.
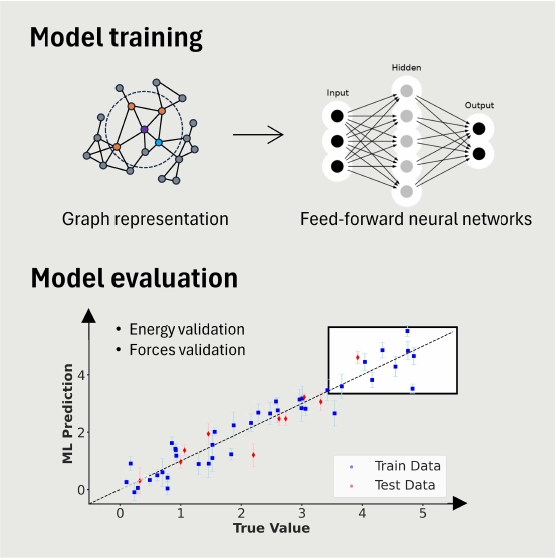
Active Learning
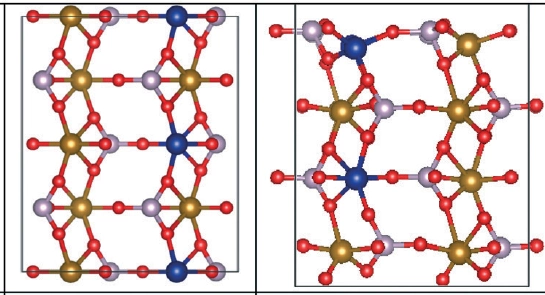
Iron-phosphate Surface
We modeled a series of transition-metal-doped Fe1−xMxCo(PO4)4(010) and Fe3Co1−xMx(PO4)4(010) electro-catalyst surfaces (with M = Mn, Os, Ru, Rh, and Ir) using density-functional theory (DFT) to evaluate their potential for OER in water-splitting processes. The DFT calculations were used to systematically analyze their electronic and thermodynamic energetics to assess OER viability, including the charge-density effects of the dopants. All OER reaction steps were explored on possible surface sites, with energy profiles and free-energy changes computed. Notably, iron replaced by osmium showed the most promising catalytic effect at Co surface sites.
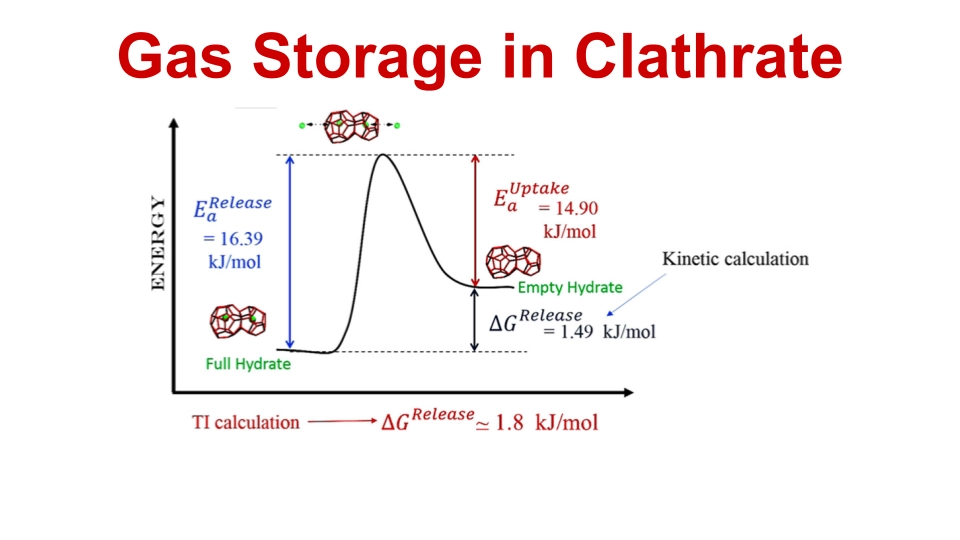
3. Electric-Field Control of Neon Uptake and Release to and from Clathrate Hydrates
We evaluated the thermodynamic and kinetic properties of neon uptake and release in a sII clathrate hydrate using molecular dynamics simulation, both with and without an applied static electric field. Neon "leakage" from the clathrate into a vacuum was simulated at temperatures from 200 to 225 K for 0.5 μs, observing progressive emptying of cages. Activation energies for release and uptake were calculated and compared with experimental values. Additionally, in the presence electric field, the release activation energy decreased gradually, indicating potentials for controlled gas release from pre-loaded clathrates.
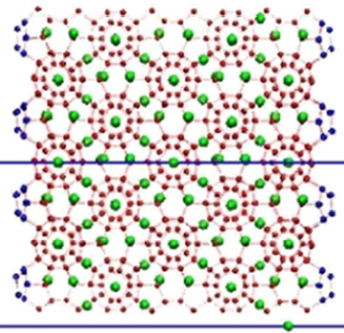
Clathrate Hydrates
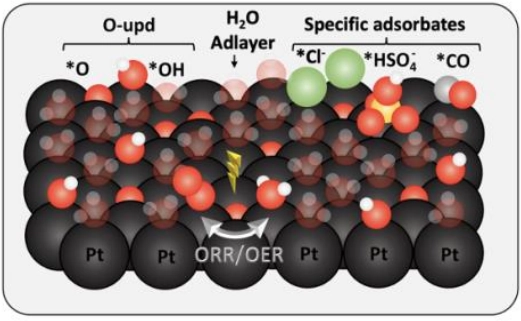
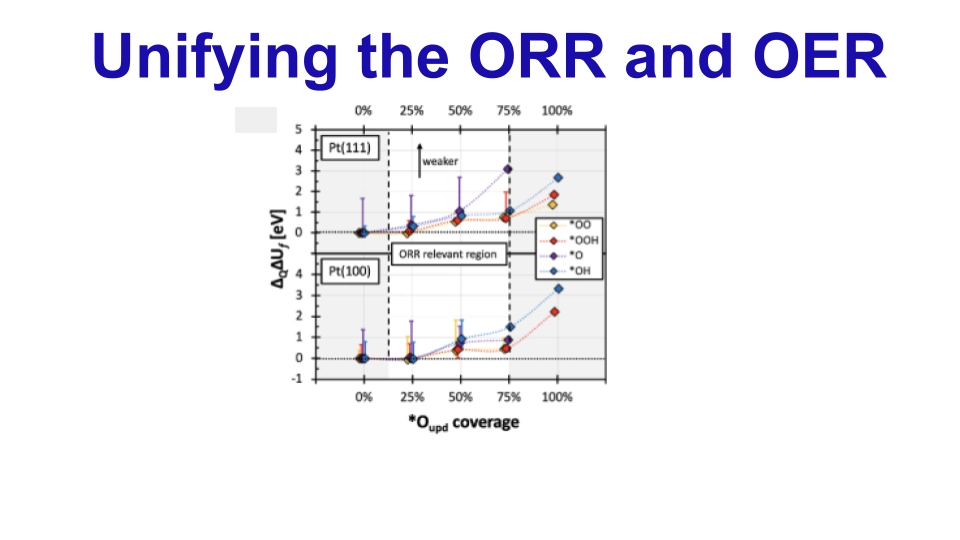
OER/ORR on Pt surface
Understanding the kinetics of the ORR is crucial in catalyst development for hydrogen fuel cells. However, traditional half-cell rotating disk electrode (RDE) protocols have limitations, as ORR activities are influenced by scan rate and surface oxygen presence. This study introduces a novel deconvolution approach to separate intrinsic ORR kinetics from surface oxygen effects on platinum catalysts, enabling precise evaluation regardless of scan rate or direction. Notably, by employing density functional theory (DFT) calculations, the study shed light on the role of surface oxygen in modulating ORR kinetics, offering valuable insights for catalyst design and fuel cell enhancement.
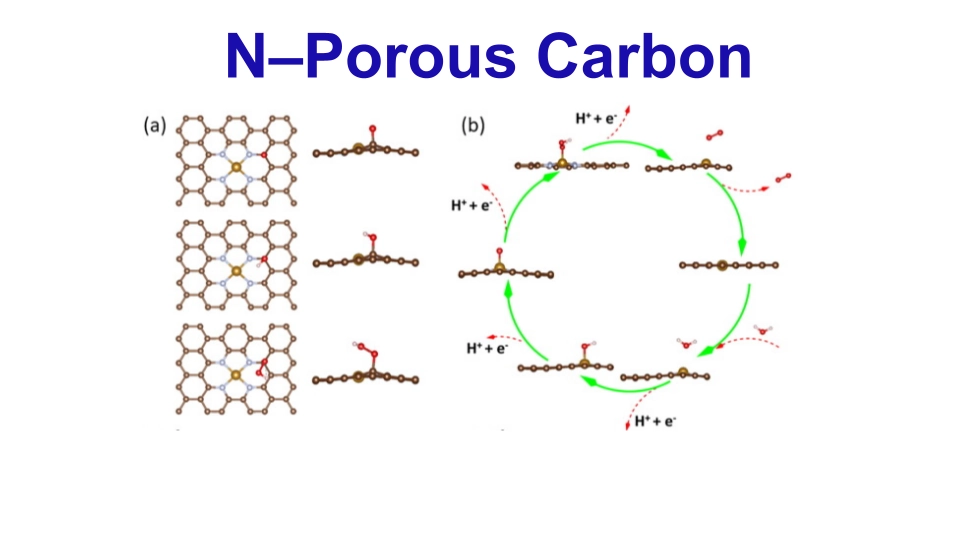
Novel electrocatalysts, which are essential for developing energy technologies, are made using a simple two-step process. Through the introduction of a heteroatom (N) and ultralow doping (0.06 wt%, Fe), defects are created, hence increasing catalytic activity. Both the hydrogen evolution process (HER) and the oxygen evolution reaction (OER) are enhanced by these defects, which form active sites inside porous channels. The experimental results demonstrate a notable improvement in the kinetics of the reaction, leading to remarkable Tafel and OER overpotential values. Carbon matrix flaws are discovered to optimize the adsorption-desorption process of OER intermediates, a finding corroborated by DFT studies and presenting a possible path toward the development of effective electrocatalysts.
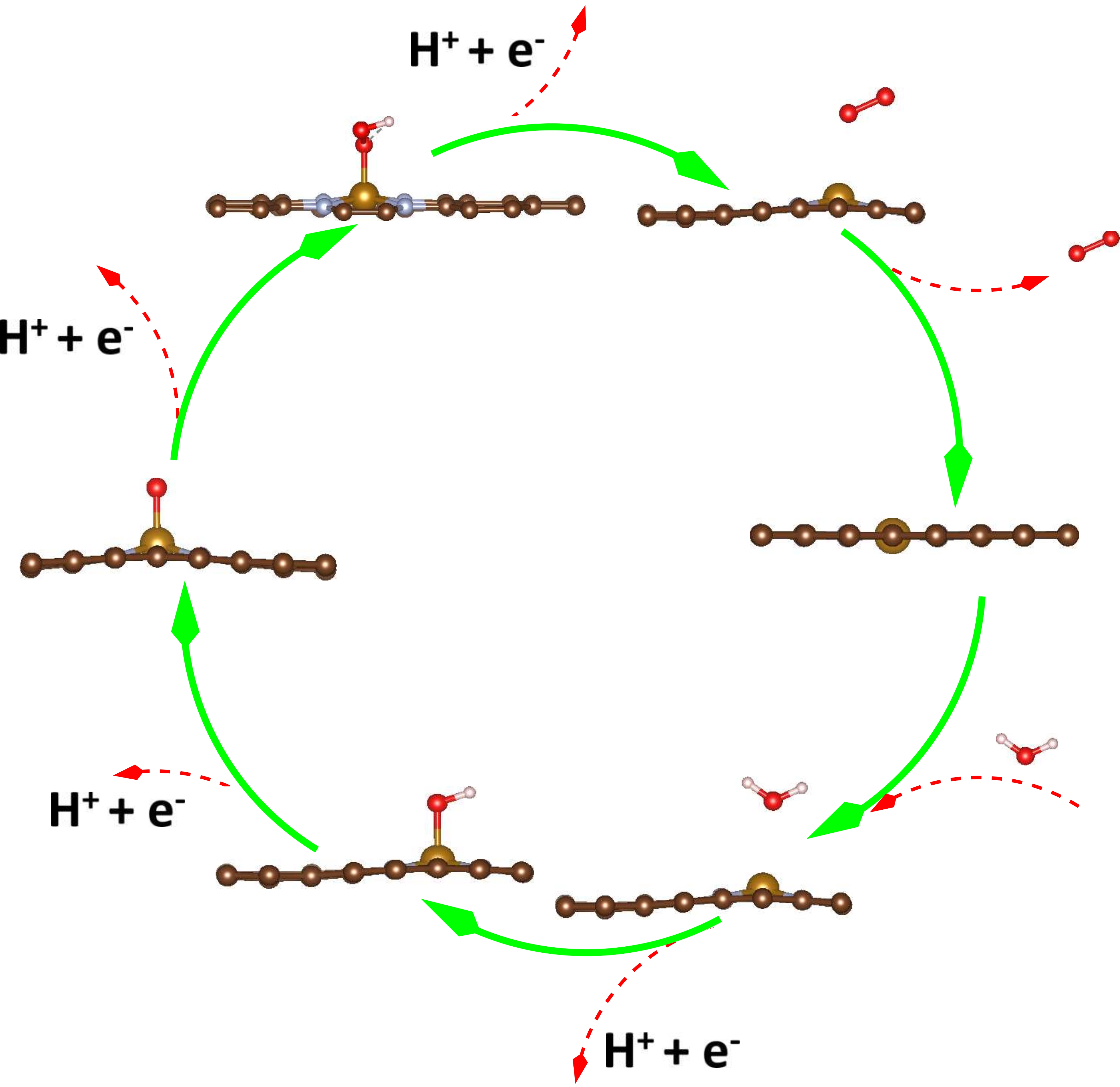
OER on Fe–N–CHPS
6. Classical Dynamics Simulations of Dissociation of Protonated Tryptophan in the Gas Phase
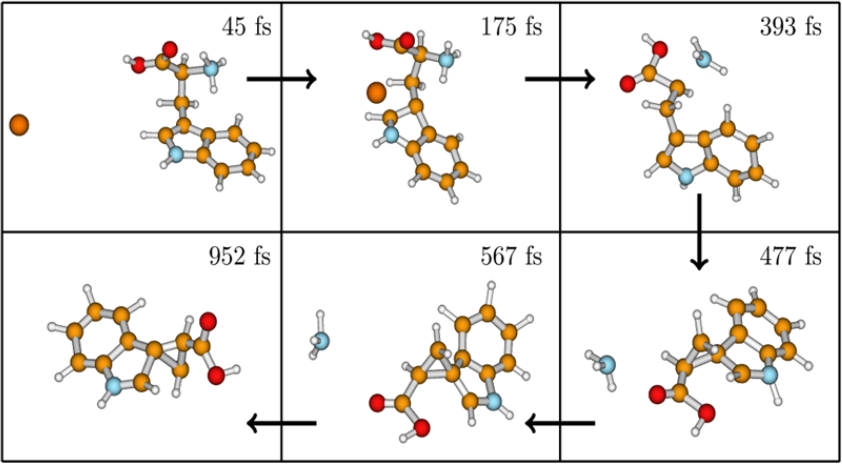
C3 nucleophilic attack
Gas phase decomposition of protonated amino acids, particularly aromatic ones like tryptophan, is crucial for understanding protein and peptide chemistry. This study employs on-the-fly classical chemical dynamics simulations with density functional theory to investigate the decomposition of protonated tryptophan. Unlike aliphatic amino acids, protonated tryptophan exhibits NH3 elimination rather than iminium ion formation, with a major fragmentation pathway involving Cα−Cβ bond fission. Results from this study align well with experiments, providing detailed atomic-level mechanisms for the decomposition process.
For more research publications, please visit my profiles on Google Scholar and ORCiD.